Project name: project-gigas_ploidy
Funding source: unknown
Species: Crassostrea gigas
variable: ploidy, desiccation, high temperature
Background
After running methylkit to find the DML within each dataset, we now need to see where they are located. To do this I will be running this jupyter notebook and using the genome feature tracks available here
Genome feature analysis
Here I use the output of getMethylDiff from our prior MethylKit analysis to create BEDfiles:
%%bash
#Replace , with tabs
#Remove extraneous quotes entries (can also be done in R)
#Print chr, start, end, meth.diff
#Remove header
#Save as BEDfile
for f in /home/mngeorge/project-gigas_ploidy/bisulfide_analysis/WGBS/DML/DML-getMethylDiff-Cov10-20-Ronit-COMBO.csv
do
tr "," "\t" < ${f} \
| tr -d '"' \
| awk '{print $2"\t"$3"\t"$4"\t"$8}' \
| tail -n+2 \
> ${f}.bed
done
Next, I used intersectBed to calculate the overlap between the getMethylDiff bed files and the genome feature track files:
#Find overlaps between DML and each genome feature - Ronit dataset, factor: ploidy
#Count number of overlaps
!intersectBed \
-u \
-a DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY.bed \
-b GFF/cgigas_uk_roslin_v1_gene.gff \
> DML/RONIT/DML-getMethylDiff-Cov10-20-gene-Ronit-PLOIDY.bed
!wc -l DML/RONIT/DML-getMethylDiff-Cov10-20-gene-Ronit-PLOIDY.bed
!intersectBed \
-u \
-a DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY.bed \
-b GFF/cgigas_uk_roslin_v1_exon.gff \
> DML/RONIT/DML-getMethylDiff-Cov10-20-exon-Ronit-PLOIDY.bed
!wc -l DML/RONIT/DML-getMethylDiff-Cov10-20-exon-Ronit-PLOIDY.bed
!intersectBed \
-u \
-a DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY.bed \
-b GFF/cgigas_uk_roslin_v1_exonUTR.gff \
> DML/RONIT/DML-getMethylDiff-Cov10-20-exonUTR-Ronit-PLOIDY.bed
!wc -l DML/RONIT/DML-getMethylDiff-Cov10-20-exonUTR-Ronit-PLOIDY.bed
!intersectBed \
-u \
-a DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY.bed \
-b GFF/cgigas_uk_roslin_v1_intron.bed \
> DML/RONIT/DML-getMethylDiff-Cov10-20-intron-Ronit-PLOIDY.bed
!wc -l DML/RONIT/DML-getMethylDiff-Cov10-20-intron-Ronit-PLOIDY.bed
!intersectBed \
-u \
-a DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY.bed \
-b GFF/cgigas_uk_roslin_v1_lncRNA.gff \
> DML/RONIT/DML-getMethylDiff-Cov10-20-lncRNA-Ronit-PLOIDY.bed
!wc -l DML/RONIT/DML-getMethylDiff-Cov10-20-lncRNA-Ronit-PLOIDY.bed
!intersectBed \
-u \
-a DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY.bed \
-b GFF/cgigas_uk_roslin_v1_mRNA.gff \
> DML/RONIT/DML-getMethylDiff-Cov10-20-mRNA-Ronit-PLOIDY.bed
!wc -l DML/RONIT/DML-getMethylDiff-Cov10-20-mRNA-Ronit-PLOIDY.bed
!intersectBed \
-u \
-a DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY.bed \
-b GFF/cgigas_uk_roslin_v1_CDS.gff \
> DML/RONIT/DML-getMethylDiff-Cov10-20-CDS-Ronit-PLOIDY.bed
!wc -l DML/RONIT/DML-getMethylDiff-Cov10-20-CDS-Ronit-PLOIDY.bed
!intersectBed \
-u \
-a DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY.bed \
-b GFF/cgigas_uk_roslin_v1_nonCDS.bed \
> DML/RONIT/DML-getMethylDiff-Cov10-20-nonCDS-Ronit-PLOIDY.bed
!wc -l DML/RONIT/DML-getMethylDiff-Cov10-20-nonCDS-Ronit-PLOIDY.bed
!intersectBed \
-u \
-a DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY.bed \
-b GFF/cgigas_uk_roslin_v1_intergenic.bed \
> DML-getMethylDiff-Cov10-20-intergenic-Ronit-PLOIDY.bed
!wc -l DML-getMethylDiff-Cov10-20-intergenic-Ronit-PLOIDY.bed
!intersectBed \
-u \
-a DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY.bed \
-b GFF/cgigas_uk_roslin_v1_rm.te.bed \
> DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY-rm.te.bed
!wc -l DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY-rm.te.bed
!intersectBed \
-u \
-a DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY.bed \
-b GFF/cgigas_uk_roslin_v1_upstream.gff \
> DML/RONIT/DML-getMethylDiff-Cov10-20-upstream-Ronit-PLOIDY.bed
!wc -l DML/RONIT/DML-getMethylDiff-Cov10-20-upstream-Ronit-PLOIDY.bed
!intersectBed \
-u \
-a DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY.bed \
-b GFF/cgigas_uk_roslin_v1_downstream.gff \
> DML/RONIT/DML-getMethylDiff-Cov10-20-downstream-Ronit-PLOIDY.bed
!wc -l DML/RONIT/DML-getMethylDiff-Cov10-20-downstream-Ronit-PLOIDY.bed
!intersectBed \
-u \
-a DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY.bed \
-b GFF/cgigas_uk_roslin_v1_flanks.gff \
> DML/RONIT/DML-getMethylDiff-Cov10-20-flanks-Ronit-PLOIDY.bed
!wc -l DML/RONIT/DML-getMethylDiff-Cov10-20-flanks-Ronit-PLOIDY.bed
Here is the output:
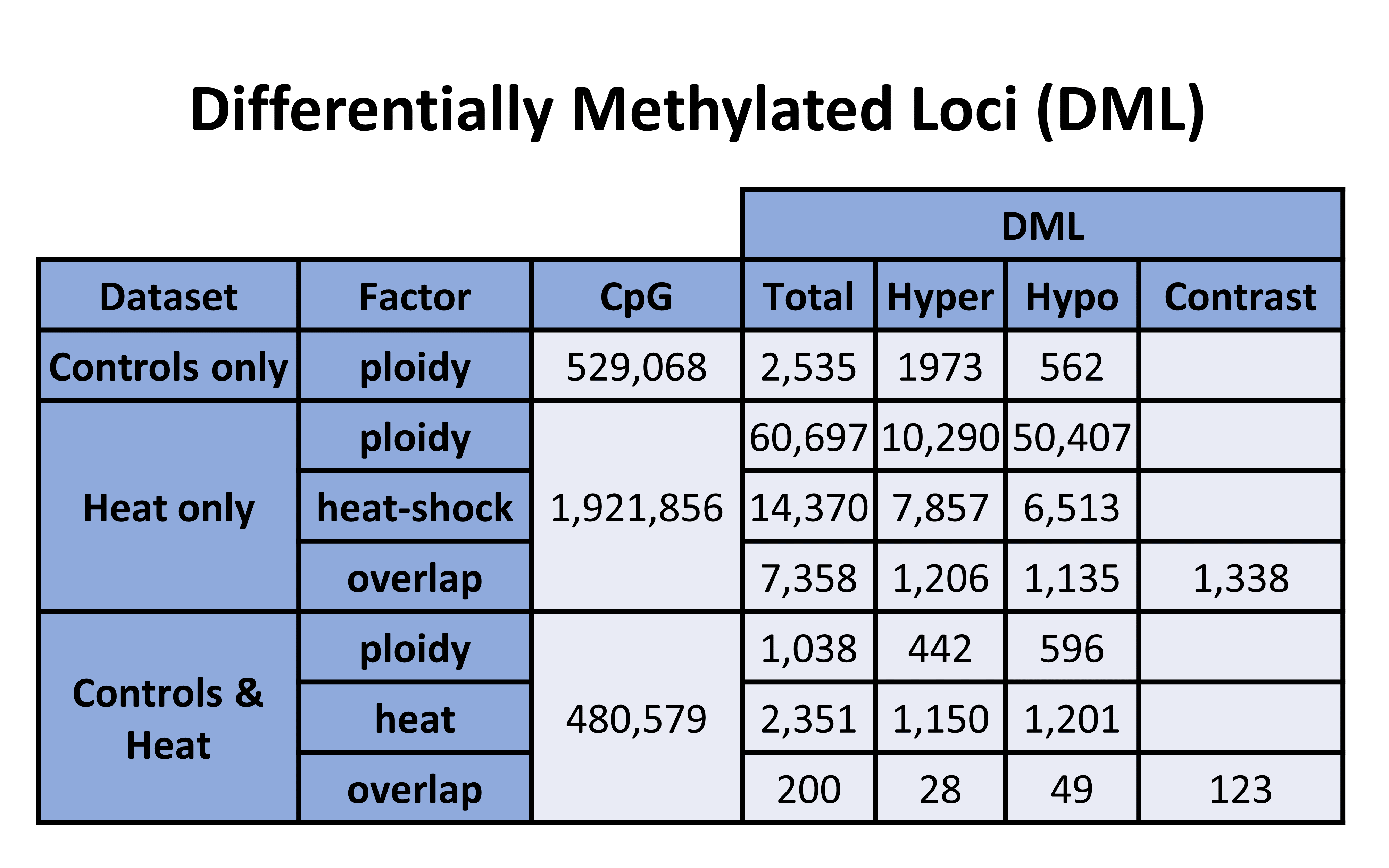
57267 DML/RONIT/DML-getMethylDiff-Cov10-20-gene-Ronit-PLOIDY.bed
19069 DML/RONIT/DML-getMethylDiff-Cov10-20-exon-Ronit-PLOIDY.bed
5416 DML/RONIT/DML-getMethylDiff-Cov10-20-exonUTR-Ronit-PLOIDY.bed
38267 DML/RONIT/DML-getMethylDiff-Cov10-20-intron-Ronit-PLOIDY.bed
1951 DML/RONIT/DML-getMethylDiff-Cov10-20-lncRNA-Ronit-PLOIDY.bed
56319 DML/RONIT/DML-getMethylDiff-Cov10-20-mRNA-Ronit-PLOIDY.bed
13654 DML/RONIT/DML-getMethylDiff-Cov10-20-CDS-Ronit-PLOIDY.bed
41696 DML/RONIT/DML-getMethylDiff-Cov10-20-nonCDS-Ronit-PLOIDY.bed
2223 DML-getMethylDiff-Cov10-20-intergenic-Ronit-PLOIDY.bed
7564 DML/RONIT/DML-getMethylDiff-Cov10-20-Ronit-PLOIDY-rm.te.bed
343 DML/RONIT/DML-getMethylDiff-Cov10-20-upstream-Ronit-PLOIDY.bed
935 DML/RONIT/DML-getMethylDiff-Cov10-20-downstream-Ronit-PLOIDY.bed
1207 DML/RONIT/DML-getMethylDiff-Cov10-20-flanks-Ronit-PLOIDY.bed
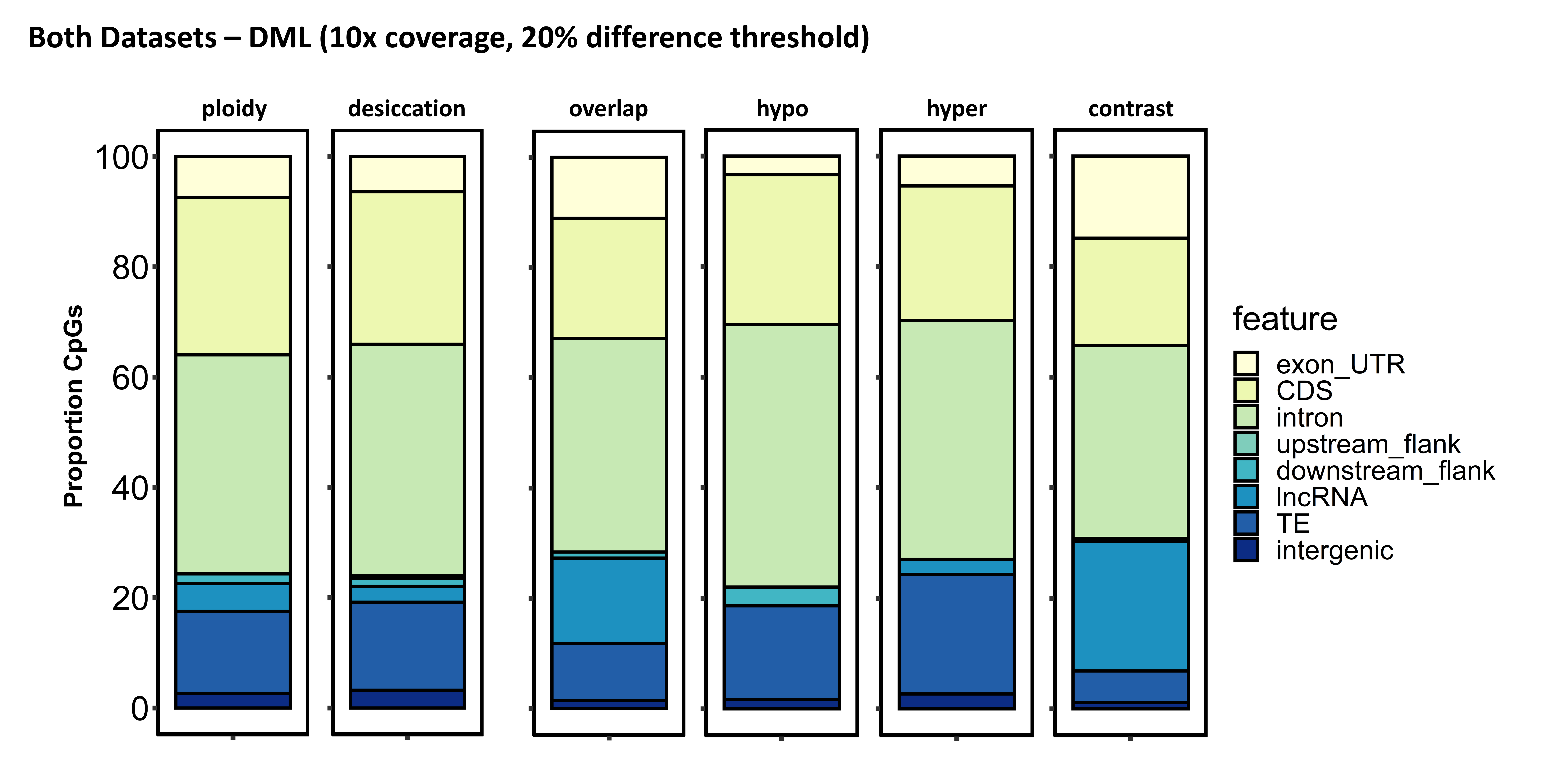
Running this code on all the outputs from all the datasets analyzed, I generated stacked bar plots linking at the proportion of CpG and DMLs within each genome feature, using the following R code:
#####################################################################
### DML-genome-feature-location Stacked bar plot
library(readxl)
library(ggplot2)
library(RColorBrewer)
data <- read_excel("DML_locations-all-datasets.xlsx", sheet = "plot", col_names = TRUE)
data$feature <- factor(data$feature, levels = c("exon_UTR",
"CDS",
"intron",
"upstream_flank",
"downstream_flank",
"lncRNA",
"TE",
"intergenic"), ordered=TRUE)
# Plot
p1 <- ggplot(data = data, aes(y = B_combo_contrast, x = 'X', fill = feature)) +
geom_bar(stat = "identity",
color = "black",
#fill = rep(v,length.out=18),
size = 1) +
scale_fill_brewer(palette = "YlGnBu") +
scale_y_continuous(limits = c(0,100), breaks = seq(0,100,20)) +
theme( line = element_line(size=1.5),
rect = element_rect(size=1.5),
text = element_text(size=22,color="black"),
panel.background = element_blank(),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.text.x = element_text(size=22,color="black"),
axis.text.y = element_text(size=22,color="black"),
# axis.ticks.y = element_blank(),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
axis.line = element_line(color = "black", size = 0.5),
panel.border = element_rect(color = "black", fill=NA, size=2))
p1
ggsave("B_combo_contrast.png",
plot = p1,
dpi = 1200,
device = "png",
width = 4.8,
height = 6,
units = "in")
##########################################################################
Here is the prior table for reference:
Table 1. DML counts from each analysis
Here are the plots:
Figure 1. Each dataset separately
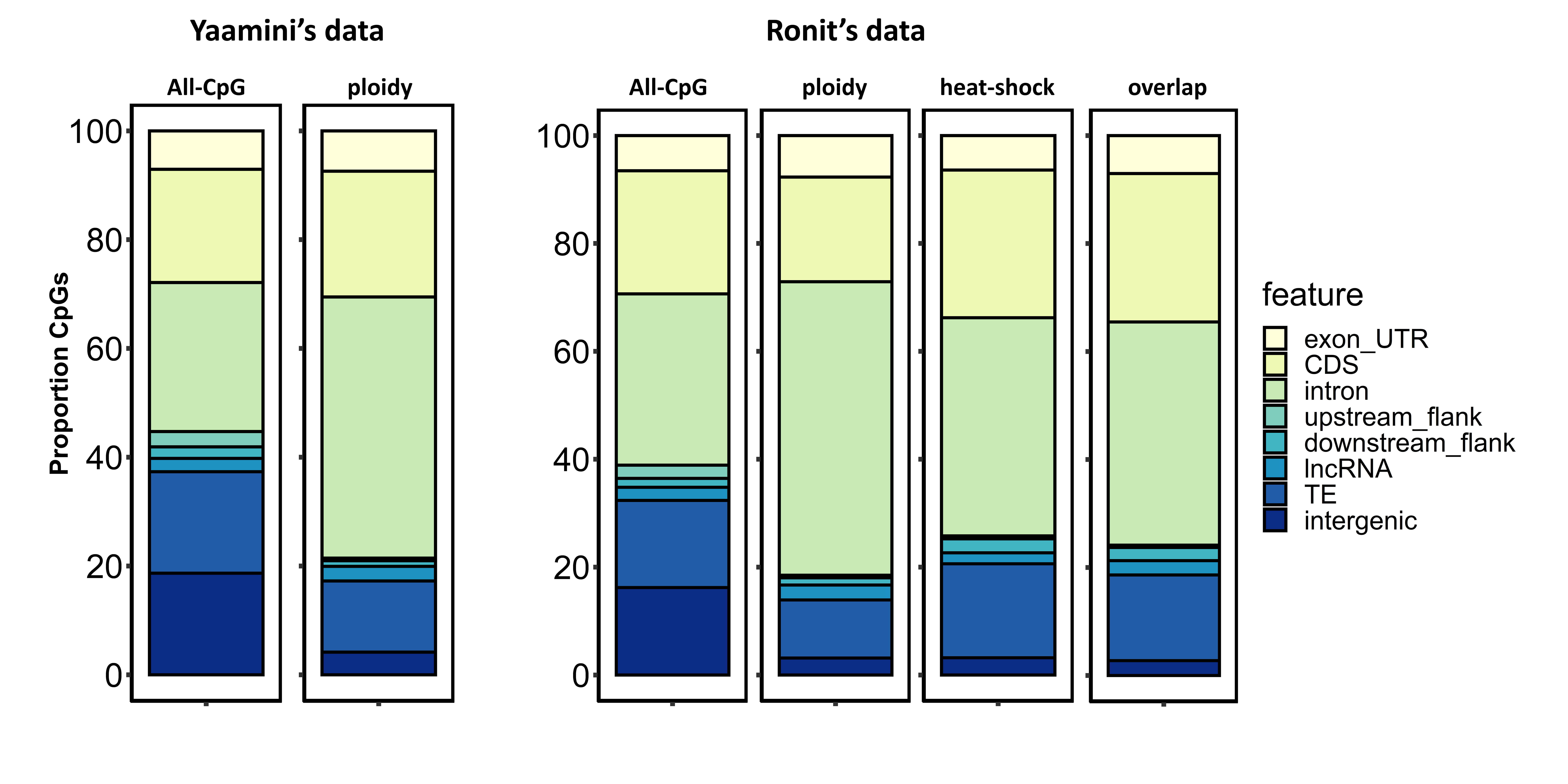
Figure 2. Both datasets together