Project name: gigas-WGBS-ploidy-desiccation
Funding source: unknown
Species: Crassostrea gigas
variable: ploidy, desiccation, high temperature
Background:
We have bisulfide sequencing data from Ronit’s desiccation exposure experiments using juvenile pacific oysters. Here is the forked github repo.
List of the progress so far:
- Received WGBS data from ZymoResearch
- Files were added to the owl server
- Sam ran FastQC. Here is the multiQC report
Here is a list of samples
| SeqID | Library Name | Tissue | Ploidy | Desiccation | Heat Stress |
|---|---|---|---|---|---|
| zr3534_1 | D11-C | ctenidia | diploid | yes | no |
| zr3534_2 | D12-C | ctenidia | diploid | yes | no |
| zr3534_3 | D13-C | ctenidia | diploid | yes | no |
| zr3534_4 | D19-C | ctenidia | diploid | yes | yes |
| zr3534_5 | D20-C | ctenidia | diploid | yes | yes |
| zr3534_6 | T11-C | ctenidia | triploid | yes | no |
| zr3534_7 | T12-C | ctenidia | triploid | yes | no |
| zr3534_8 | T13-C | ctenidia | triploid | yes | no |
| zr3534_9 | T19-C | ctenidia | triploid | yes | yes |
| zr3534_10 | T20-C | ctenidia | triploid | yes | yes |
Desiccation - desiccation for 24 hr at 27C,
Heat_stress - 1 hr at 45C
Pathway forward:
Now that we have the WGBS files and FastQC didn’t find any large errors, the next steps is to run Bismark. The files are pretty big, so instead of running it locally, I will use the resources of our hyak_mox server. Bismark performs alignments of bisulfite-treated reads to a reference genome and cytosine methylation calls at the same time.
The steps I will be following during this analysis are:
- logging into mox server
- Generating slurm script (.sh)
Step 1: Logging into mox
ssh mngeorge@mox.hyak.uw.edu
Step 2: slurm script and job scheduler
Slurm is an open source, fault-tolerant, and highly scalable cluster management and job scheduling system for large and small Linux clusters. Useful information can be found in the wiki and this example that Steven provided. Yaamini has also outlined a great pipeline for analysis and reporting.
To configure the job, I first had to develop my file structure:
mkdir -p mneorge/{analyses,blastdb,data,jobs,programs,sbatch_scripts}
parent folder: /gscratch/srlab/mngeorge
contents: analyses, blastdb, data, jobs, programs, sbatch_scripts
After completed, I copied ronit’s data to my data folder:
cp -avr /gscratch/srlab/sr320/data/cg /gscratch/srlab/mngeorge/data/cgigas_ronit
I then recreated a sbatch file within the sbatch_scripts (20210316_cgig_ploidy_stress_bismark.sh) subfolder to run bismark on the data.
Here is the code:
GNU nano 2.3.1 File: 20210316_cgig_ploidy_stress_bismark.sh
#!/bin/bash
## Job Name
#SBATCH --job-name=ronit-bismark
## Allocation Definition
#SBATCH --account=srlab
#SBATCH --partition=srlab
## Nodes
#SBATCH --nodes=1
## Walltime (days-hours:minutes:seconds format)
#SBATCH --time=15-00:00:00
## Memory per node
#SBATCH --mem=100G
#SBATCH --mail-type=ALL
#SBATCH --mail-user=mngeorge@uw.edu
## Specify the working directory for this job
#SBATCH --chdir=/gscratch/srlab/mngeorge/20210316-cgigas-ploidy-stress-bismark/
# Directories and programs
bismark_dir="/gscratch/srlab/programs/Bismark-0.21.0"
bowtie2_dir="/gscratch/srlab/programs/bowtie2-2.3.4.1-linux-x86_64/"
samtools="/gscratch/srlab/programs/samtools-1.9/samtools"
reads_dir="/gscratch/mngeorge/data/cgigas_ronit"
genome_folder="/gscratch/srlab/sr320/data/Cgig-genome/roslin_M/"
source /gscratch/srlab/mngeorge/sbatch_scripts/20210316_cgig_ploidy_stress_bismark.sh
${bismark_dir}/bismark_genome_preparation \
--verbose \
--parallel 28 \
--path_to_aligner ${bowtie2_dir} \
${genome_folder}
# /zr3644_11_R2.fastp-trim.20201206.fq.gz
find ${reads_dir}*_R1.fastp-trim.20201202.fq.gz \
| xargs basename -s _R1.fastp-trim.20201202.fq.gz | xargs -I{} ${bismark_dir}/bismark \
--path_to_bowtie ${bowtie2_dir} \
-genome ${genome_folder} \
-p 8 \
-score_min L,0,-0.6 \
--non_directional \
-1 ${reads_dir}{}_R1.fastp-trim.20201202.fq.gz \
-2 ${reads_dir}{}_R2.fastp-trim.20201202.fq.gz \
find *.bam | \
xargs basename -s .bam | \
xargs -I{} ${bismark_dir}/deduplicate_bismark \
--bam \
--paired \
{}.bam
${bismark_dir}/bismark_methylation_extractor \
--bedGraph --counts --scaffolds \
--multicore 28 \
--buffer_size 75% \
*deduplicated.bam
# Bismark processing report
${bismark_dir}/bismark2report
#Bismark summary report
${bismark_dir}/bismark2summary
#run multiqc
/gscratch/srlab/programs/anaconda3/bin/multiqc .
# Sort files for methylkit and IGV
find *deduplicated.bam | \
xargs basename -s .bam | \
xargs -I{} ${samtools} \
sort --threads 28 {}.bam \
After saving, I then added to the queue:
sbatch 20210316_cgig_ploidy_stress_bismark.sh
You can check the position in the queue using squeue:
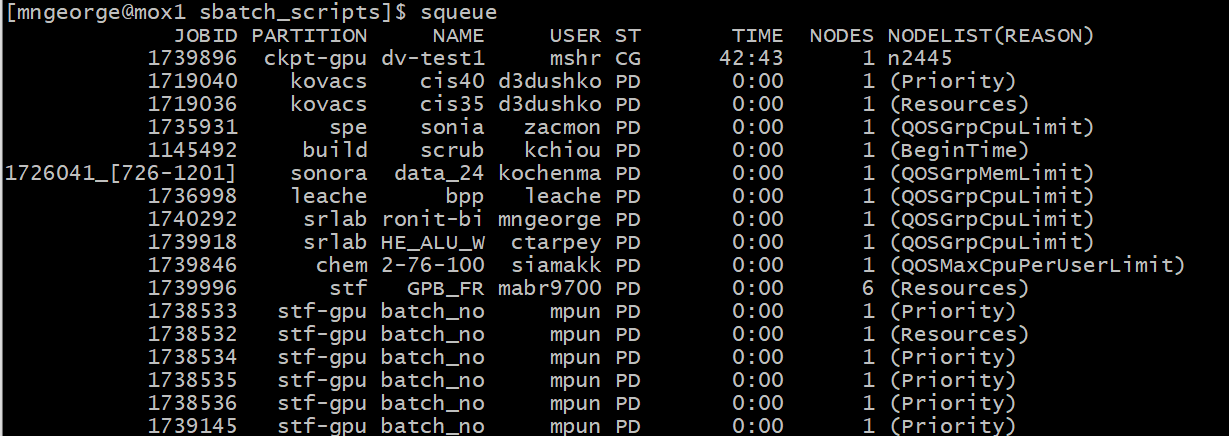
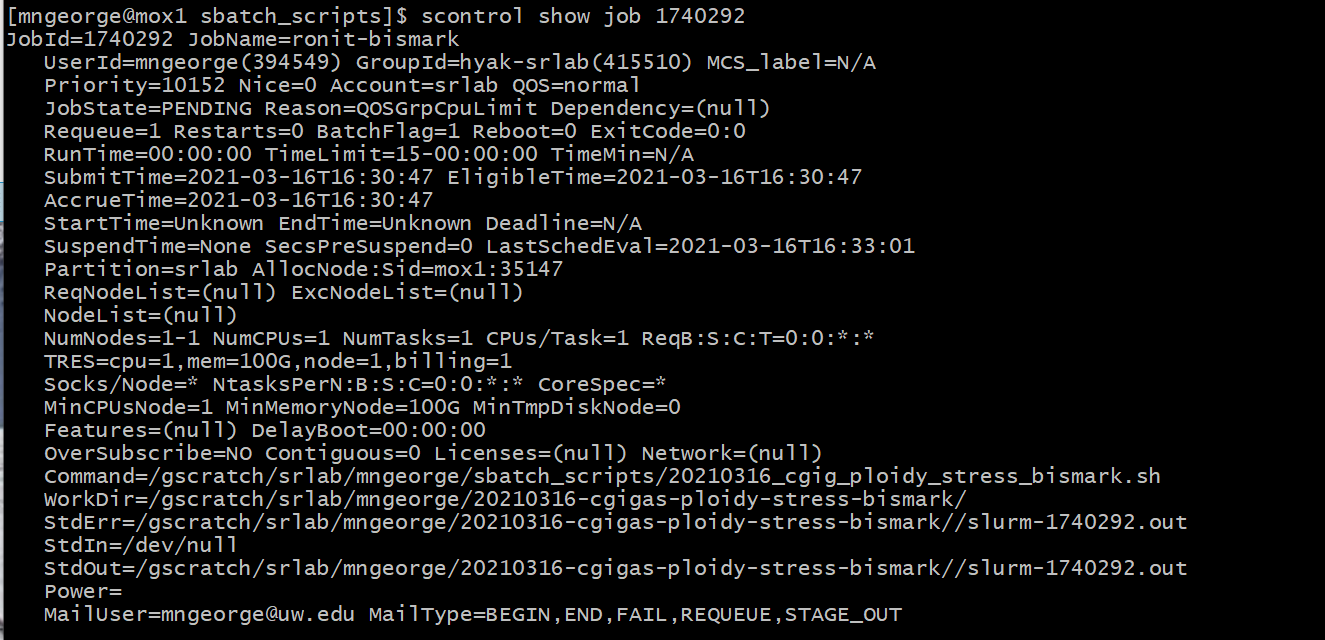
As well as the job status using the job number:
scontrol show job 1740292
Once the job is done, the resulting files should be stored on our lab server gannet. Here is an example script using rsync:
ssh mngeorge@gannet.fish.washington.edu <login>
cd /volume2/web/panopea/030521-ronrosM/
rsync -avz --progress --verbose mngeorge@mox.hyak.uw.edu:/gscratch/scrubbed/sr320/030521-ronrosM 030521-ronrosM