Project name: USDA-NRSP-8-gigas-rDNA
Funding source: USDA-NRSP-8
Github repo: https://github.com/mattgeorgephd/USDA-NRSP-8-gigas-rDNA
Species: crassostrea gigas
variable: ploidy
« previous notebook entry « | » next notebook entry »
DNA extraction and prep
Four oyster families (F05, F12, F13, & F14) were generated by Pacific Hybreed. Triploid oysters were made from each family by crossing a tetraploid with a diploid, generating half-sibling diploid and triploids within each family. Oyster spat was transferred to the Jamestown Pt. Whitney Shellfish Hatchery on 07-28-2022. Oysters were placed in heath trays and fed a mixed algae diet ad libitum. Whole body oyster tissue samples were collected on 11-14-2022; 15 samples were collected for each ploidy within each family. Samples were stored in the FTR -80 at position 5-1-3 & 5-1-4.
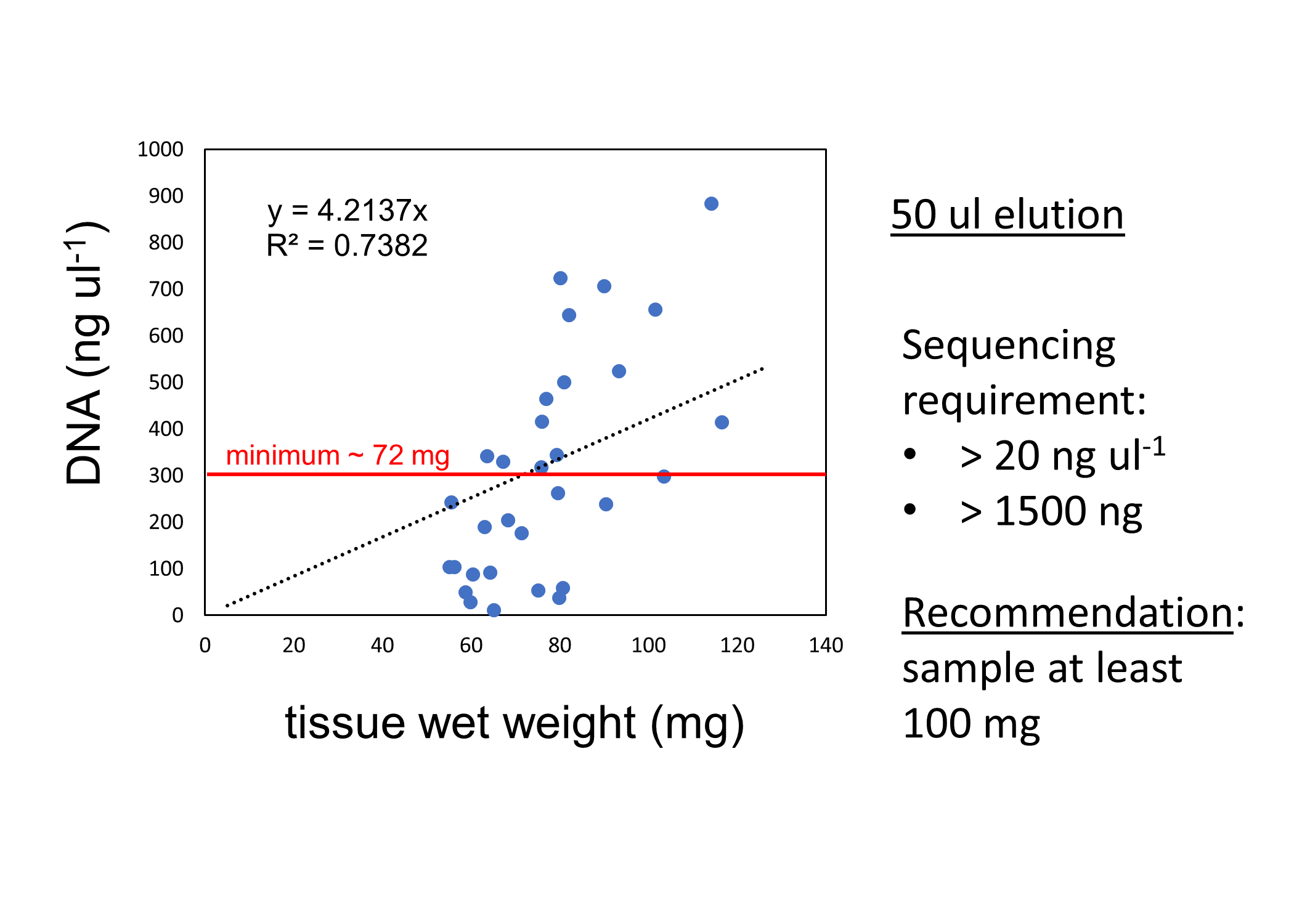
DNA was extracted from diploid and triploid juvenile Pacific oysters from two families (F05 and F14). We used the Omega Bio-Tek E.Z.N.A.® Mollusc DNA Kit; here is our protocol. We found that at least 100 mg of tissue was needed to get the amount of DNA needed to submit for Azenta for sequencing (assuming elution volume of 50 ul: at least 20 ng ul-1; at least 1500 ng total).
Grace Leuchtenberger (BIO Grad Student) and Henry Berg (SAFS undergraduate) helped with extractions. Here is the DNA extraction datasheet with sample manifest.
DNA Sequencing
We submitted 32 samples from two families to GeneWiz (Azneta) for sequencing on February 28. Here is the breakdown:
| Family | Ploidy | sample_ID |
|---|---|---|
| F05 | diploid | F052n01 |
| F05 | diploid | F052n02 |
| F05 | diploid | F052n03 |
| F05 | diploid | F052n04 |
| F05 | diploid | F052n05 |
| F05 | diploid | F052n06 |
| F05 | diploid | F052n07 |
| F05 | diploid | F052n08 |
| F05 | triploid | F053n01 |
| F05 | triploid | F053n02 |
| F05 | triploid | F053n03 |
| F05 | triploid | F053n04 |
| F05 | triploid | F053n05 |
| F05 | triploid | F053n06 |
Here is the link to the Azenta Quote. We sequenced at 30x coverage. It was assigned project number 30-835022638.
We Received the data on back on 04-29-2023. Sequencing data was provided as zipped fastq files on the genewiz sFTP server. Here are the server details:
Host: sftp://sftp.genewiz.com
User: mngeorge_uw
Password: pxLSUtDDLhprLvLkweVf
Port: 22
The Genewiz download guide is provided here. I downloaded the sequencing files directly to our lab sequencing repository “nightingales” on owl. Here is link to the C_gigas folder. Here is the code used to transfer:
# Login to owl
ssh mngeorge@owl.fish.washington.edu
# Set local working directory where files will be transferred to
cd /var/services/web/nightingales/C_gigas
# Login to sFTP server
sftp mngeorge_uw@sftp.genewiz.com
# change folder to Genewiz project folder w/ fastq files
cd 30-835022638/00_fastq
# confirm local dir
lpwd
# confirm remote working dir
pwd
# Transfer files from sFTP to Owl
mget *
Sequence QC, trimming
I downloaded the sequence files onto Raven in the “data/raw” using the same sftp method as described above. The next step was to unzip the fastq files:
# unzip .fastq.gz files
cd data/raw/
gunzip *.fastq.gz
Aand then run fastqc and multiqc on the on the raw data
Run fastqc on untrimmed files
mkdir fastqc/
mkdir fastqc/untrimmed/
/home/shared/FastQC/fastqc \
data/raw/*.fastq \
--outdir fastqc/untrimmed/ \
--quiet
Run multiqc on untrimmed files
eval "$(/opt/anaconda/anaconda3/bin/conda shell.bash hook)"
conda activate
cd fastqc/untrimmed/
multiqc .
The multiqc report for the raw data is here
I then trimmed adapter sequences (hard trimmed first 10 bps):
# trim adapter sequences
mkdir data/trimmed/
cd data/raw/
for F in *.fastq
do
#strip .fastq and directory structure from each file, then
# add suffice .trim to create output name for each file
results_file="$(basename -a $F | sed 's/\.[^.]*$/_trim&/')"
# run cutadapt on each file, hard trim first 10 bp
/home/shared/8TB_HDD_02/mattgeorgephd/.local/bin/cutadapt $F -u 10 -o \
/home/shared/8TB_HDD_02/mattgeorgephd/USDA-NRSP-8-gigas-rDNA/data/trimmed/$results_file
done
and concatenated the fastq files by sequencing run
mkdir data/trim-merge/
# Set the input and output directories
input_dir="/home/shared/8TB_HDD_02/mattgeorgephd/USDA-NRSP-8-gigas-rDNA/data/trimmed"
output_dir="/home/shared/8TB_HDD_02/mattgeorgephd/USDA-NRSP-8-gigas-rDNA/data/trim-merge/"
# Loop through all of the R1 fastq files in the input directory
for r1_file in "$input_dir"/*_R1_*.fastq
do
# Extract the sequencing run from the R1 file name
run=$(basename "$r1_file" | cut -d'_' -f1,2)
# Find the corresponding R2 file
r2_file="$input_dir"/"${run}_R2_*.fastq"
# Concatenate the R1 and R2 files and save the output to a new file in the output directory
cat "$r1_file" "$r2_file" > "$output_dir"/"${run}_trim-merge.fastq"
done
and again run fastqc and multiqc
Run fastqc on trimmed & merged files
mkdir fastqc/
mkdir fastqc/trim-merge/
/home/shared/FastQC/fastqc \
data/trim-merge/*.fastq \
--outdir fastqc/trim-merge/ \
--quiet
Run multiqc on trimmed & merged files
eval "$(/opt/anaconda/anaconda3/bin/conda shell.bash hook)"
conda activate
cd fastqc/trim-merge/
multiqc .
Here is the final multiqc report for the trimmed and merged files can be found here
The files are on Raven in:
/home/shared/8TB_HDD_02/mattgeorgephd/USDA-NRSP-8-gigas-rDNA/data/trim-merge